Several analytical programs developed by our group are listed under the modules
entry in the main menu. They perform several tasks providing a powerful
combination of tools to analyze the given PDB structure.
(See here how to make external links
to all STING modules)
(Also, verify here how to get to results in Warp speed!
Make extensive use of PDB_Metrics
as well!)
These modules are:
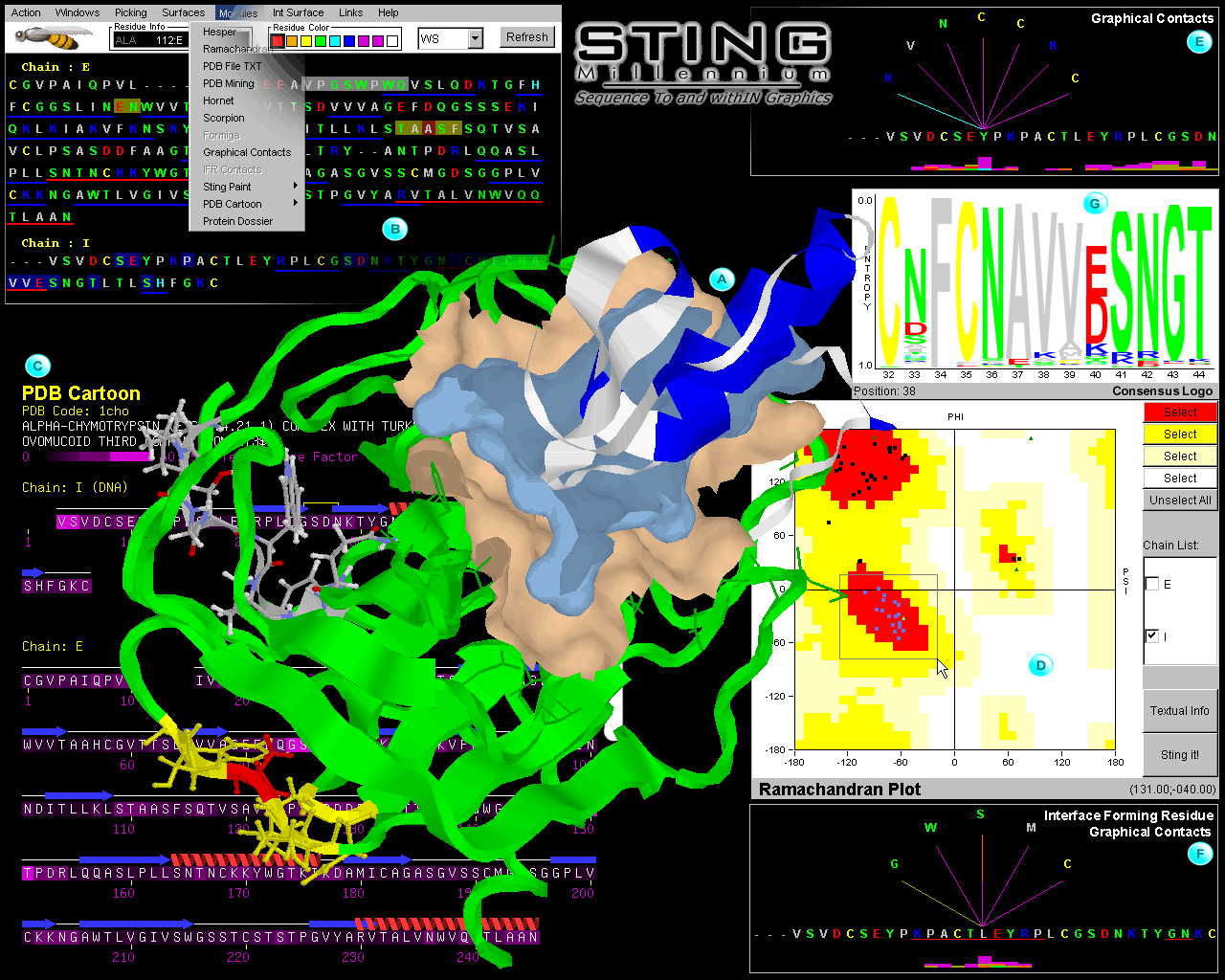
1) Ramachandran plot
(Ramachandran et al., 1963):
A fully interactive plot displaying the main-chain dihedral angles (pfi
and psi). It identifies the allowed/disallowed regions for the angles
(inset: D on fig. 1) and permits selection of residues that
can be displayed in the structure -Graphics window.
2) PDB file TXT:
Lists the original PDB entry in text format.
3) Hornet:
This module performs calculation and visualization of hydrogen bonds between
two protein chains with special attention given to water molecules (trapped
between two chains) participating in H-bond formation.
7) Graphical IFR Contacts:
IFR Contacts is relaying absolutely on the same concept as Graphical Contacts;
Here though, the contacts are only calculated among the IFRs belonging to the
facing protein chains at the interface. The user should remember that
this option is activated by initially defining the Interface.
Note that the chain which is chosen here {the IFR Contacts panel: yellow line
circled item} is used as the base (horizontal) sequence. The other chain
will contain the amino acids (in contact with the base sequence) that
would be presented on the other end of the fan-like contact graphics. The base
/central residue is color coded white in 3D. Note that the histogram showing
total number and the type of contacts, only appears here among the red underlined
residues in the base (horizontal) sequence: the IFRs.
The table of contacts on the top right inset has same features as the table
of internal contacts (see
description above). Here though, pairing amino acids belong to the facing
protein chains (therefore chains labeled with the different single letter code).
IFR contacts now includes also those residues that can make contact but do not belong to the INTERFACE! This is because Interface Forming Residues are strictly defined based on solvent accessibility. However, even those residues on the surface of facing chains that did NOT loose accessibility to the solvent, still can make for example HB, as the distance from donor to the acceptor is larger than the diameter of the water molecule used to define IFRs. See blue underlined residues (extended interface) in PD Interface (red underlined) [D_25 and N_29 in Chain 1]{in PD example under the pointer ® on the left!}. Note: as IFR Graphical Contacts does not have access to data on solvent accessibility, above mentioned residues are underlined in red as if they belong to the Interface ansamble strictu sensu!
8) StingPaint:
This is a visual aid to display multiple sequence alignments with the amino
acids colored by physico-chemical properties. STINGpaint now supports the following
sequence and Multiple Sequence Alignment (MSA) formats: Coloring sequence of
any PDB entry, Coloring any sequence in FASTA format, Coloring MSA in CE
(Ilya N. Shindyalov and Philip E. Bourne (1998)) and PRISM
(Yang, A.S. & Honig, B. (1999)) output format Coloring MSA in CLUSTAL-W,
PSI-BLAST and GCG output format.
11) ConSSeq:
This service presents PDB file sequence and consensus sequence (as found in HSSP)
colored by conservation, color coded graphic bars of relative entropy, information
about residues present in other homologous sequences, with their respective
frequency. For fast visualization, this program generates the sequence logo.
12) Java Ca-Ca/Cb-Cb Distance Plot:
Java Ca-Ca Distance Plot module displays the level curves of the distances between the alfa carbon of each residue and alfa carbons atons of every other residues, within a single chain of the PDB file.
Java CB-CB Distance Plot module displays the level curves of the distances between the beta carbon of each residue and beta carbons atons of every other residues, within a single chain of the PDB file.
Java Protein Dossier
Java Protein Dossier is a interactive presentation of important physical-chemical
characteristics of the macromolecular structure described in a PDB file. With
a few mouse clicks the user can access data about chosen parameter, call other
Blue Star Sting modules or refine the search for a specific characteristics. By
using color code scales for each residue of the sequence, JPD shows corresponding:
temperature factor, solvent accessibility of the single chain (and also in complex
with the other present chains in given PDB file), hydrophobicity, sequence conservation
in a multiple alignment (relative entropy), double occupancy, reliability and
histograms representing the atomic contacts. JPD also shows the identification
of Interface Forming Residue (IFR) and their internal contacts. JPD offers information
about electrostatic potential and curvature on protein surface. In addition,
comparison of the Secondary Structure annotated by PDB, by DSSP and by Stride
is presented.
STING Report
STING Report is a web based application for extracting concise and
focused information from the Blue Star STING Data Base on specific amino acids
within the structures described in PDB files. The extracted information is presented
to a user through a series of GIF images and a table of numerical values for
the set of structure/function descriptors/parameters, generated by the Blue Star STING components. The resulting HTML page with the GIF images and the table
is ready to be printed and most importantly, it can be visualize at platforms
with the most elementary configurations (no java, chime, plug-ins etc.). Below
is a schematic view of GIF image panels which are generally produced by the
Blue Star STING Report. We have used 1cho.pdb, chain E, residue HIS_57 to generate
Blue Star STING Report for this help. Other panels will be added with the time,
making Blue Star STING Report GIF image collection more extensive.
Java Table of
Contacts
Java Table of Contacts is a interactive tool for listing contacts between residues
of one protein and dividing them in specific classes. JTC shows all contacts
for a specified residue, IFR Contacts and also Protein-DNA Contacts. This interactive
tool allows user to access other Blue Star Sting components or have physical-chemical
parameters already "painted" within the table, such as: relative entropy,
accessibility, secondary structure and the distance for the identified contacts.
The user can make a choice of the residues important for his consideration,
make them appropriately displayed and then print the table of publication quality.
Phylogenetic
Tree
STING_P3 sands for Blue Star Sting Phylogenetic Tree component.
Phylogenetic tree is generated starting from either HSSP or SH2Qs
multiple sequence alignments. TreeView standard viewer package is used to present
to a user calculated phylogenetic trees.
Blue Star STING MSA
STING_MSA stands for the new Blue Star Sting component: Multiple
Sequence Alignment. Those sequences are extracted both from HSSP and from our
own SH2Qs (Sequences Homologue to the Query [Structure-having]
Sequence). The latter one is designed so that we can benefit from the possibility
of making a choice of the number of sequences that we want to consider in the
relative entropy calculation. STING_MSA will use standard MSA viewer package:
SeaView.
Blue Star STING DNA
Contacts
This Blue Star Sting component demonstrates relevant contacts being
established across the interface formed by protein and DNA chain. Similar logistics
is used here as in IFR Graphical Contacts.
XY Graphics Java
Plot
XY Graphics Java Plot is the STING component which permits to
a user to examine one of 150 different parameters in a simple yet intuitive
plot showing the numerical values at theY-axes and the sequence residue numbers
at the X-axis.
STING_TGZ module generates a compressed file containing all the files used by several STING's modules, for example, JPD (Java Protein Dossier). These files are saved in different directories within the compressed file. If your research project needs to analyze the same pdb file very often, the best way to save time (in downloading data from internet) is to use STING_TGZ module..
Multiple Structure - Single Parameter (MSSP) 2D plot
The Multiple Structures - Single Parameter 2D module allows the user to compare, in a graphical manner, any one of the 150 different parameters in different protein structures in a simple yet intuitive plot. The plot shows the numerical values in the Y-axis and the sequence residue numbers in the X-axis.
STING_DB is composed of structural, sequence, function and stability parameters/descriptor for protein analysis. This database operates with a collection of both publicly available data (PDB, HSSP, Prosite) and its own data (contacts, interface contacts, surface accessibility). STING_DB is one of the best known databases of structural parameters reported in per-residue fashion with over 300 of them compiled at a single site.
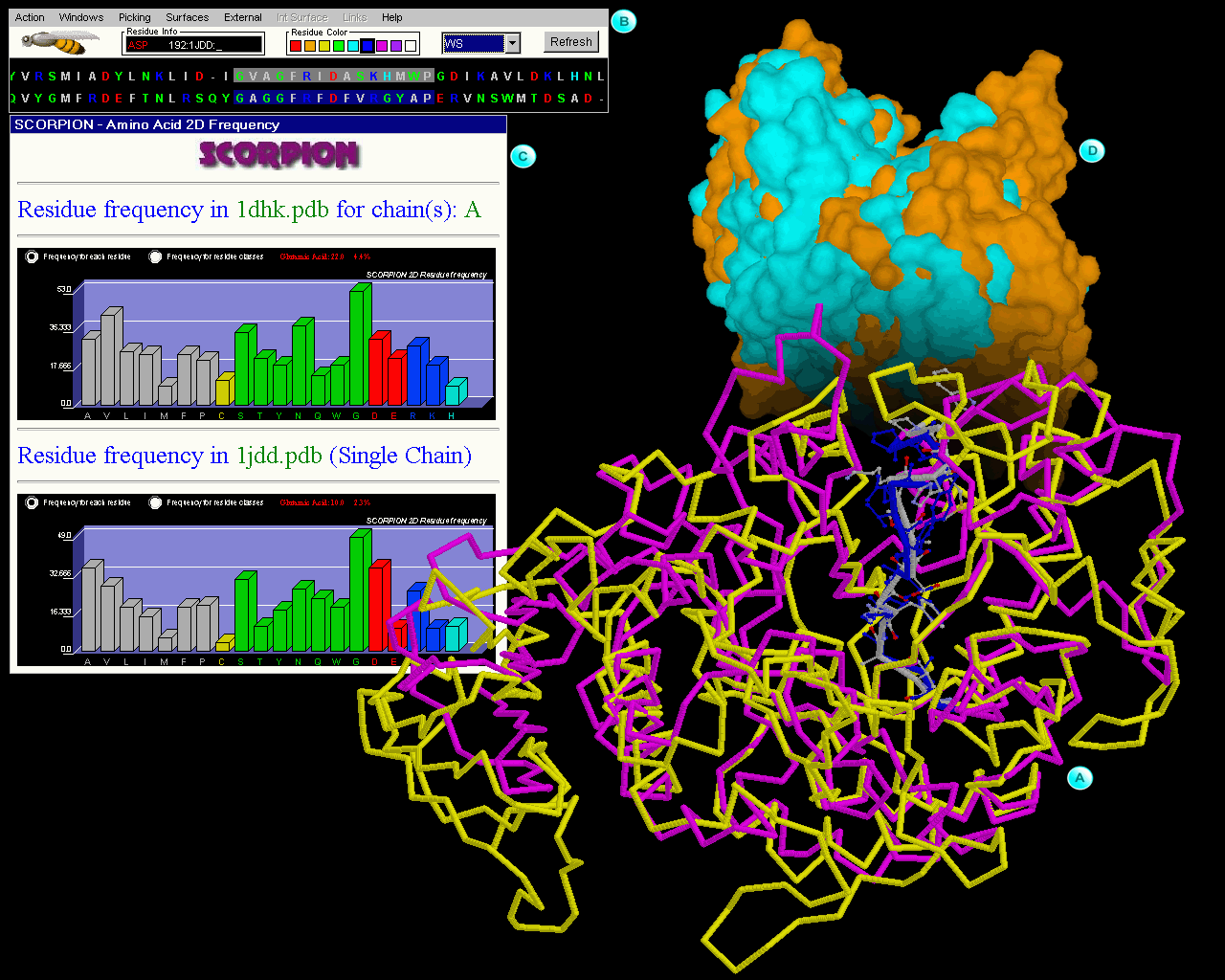
JPIV is a module of STING suite. It is designed to visualize and analyze the proteins' interfaces. In order to accomplish this, the interfaces are displayed separately, as shown in Figure 1, and over each one of them is painted a common physical-chemical property, available from STING_DB, that could be important in the complex formation. The contact interfaces are generated using weighted Delaunay triangulation and marching tetrahedra algorithms from the CGAL library, afterwards they and rendered in an Interactive Java Viewer based on JavaView library.
The "AA Co-evolution" is the STING component which permits a user to examine co-evolution parameter of residues in a simples, yet intuitive, 3D plot. A user might confirm which amino-acid positions were identified by STING, within the previously confirmed amino acid conserved set, as evolved in concert.
This STING module can analyze the interatomics contacts in protein-ligand complexes. The contacts shown by this STING module include those mediated by charge-charge interactions hydrogen bonds and hydrophobic contacts.
Contacts Distance Maps (CDM) is a STING's module. It is designed to display graphically all the protein’s contacts. Moreover, the Secondary Structure (SS) elements are also shown in a single line, facilitating theirs visualization for each residue, and contour curves distances maps involving a carbon, ß carbon and LHA-LHA (Least Heavy Atom) are presented in a Java interactive interface..
In STING_PCD users can obtain a complete comparison report of the intrachain interactions for any two chains described in the PDB format file. The user must supply any two PDB ids and corresponding chain ids for the analysis. At the output, a user will receive a list of interactions which were preserved in both chains as well as the list of those which are present in only one of them. Users can also analyse a wild type protein and a corresponding mutant structure with a single point mutantion; in this case a user needs to supply at the input, the mutated amino acid name and the number.
STING_TopSiMap is a module which makes possible to compare the contact maps of two chains in terms of the preserved interactions as well as the ones which are present in only one of the two chains analysed. Users can see / print the images of the maps and also view the contacts in a JMol window where STING presents two structurally aligned chains. Users can also compare the chains in terms of the contacts classification (hydrophobic, aromatic stacking, hydrogen bonds, salt bridges and cysteine bridges) and corresponding energies.
In this module, users can obtain the index for a toplogy similarity for a list of up to 100 structures. By submitting the STING TGZ file (containing all STING_DB parameters, precalcualated, corresponding to certain public or local structure) and a text file containing the list of up to 99 public PDB files, user can start the procedure which calculates the similarity index among selected (up to) 100 structures. This index is a reliable measure of the changes in the pattern of the intrachain interactions and can be used to analyse the topology similarities.
This is a database of homologous PDB chains based on their interactions (contacts) pattern. For each of the "ASTRAL 40" ensamble chains, we computed a list of the most similar chains based on their contact maps. The database is searchable and for each chain, the list of the topologs is presented.
Under construction.
| Fig_1. |
|
Fig_2. |
|