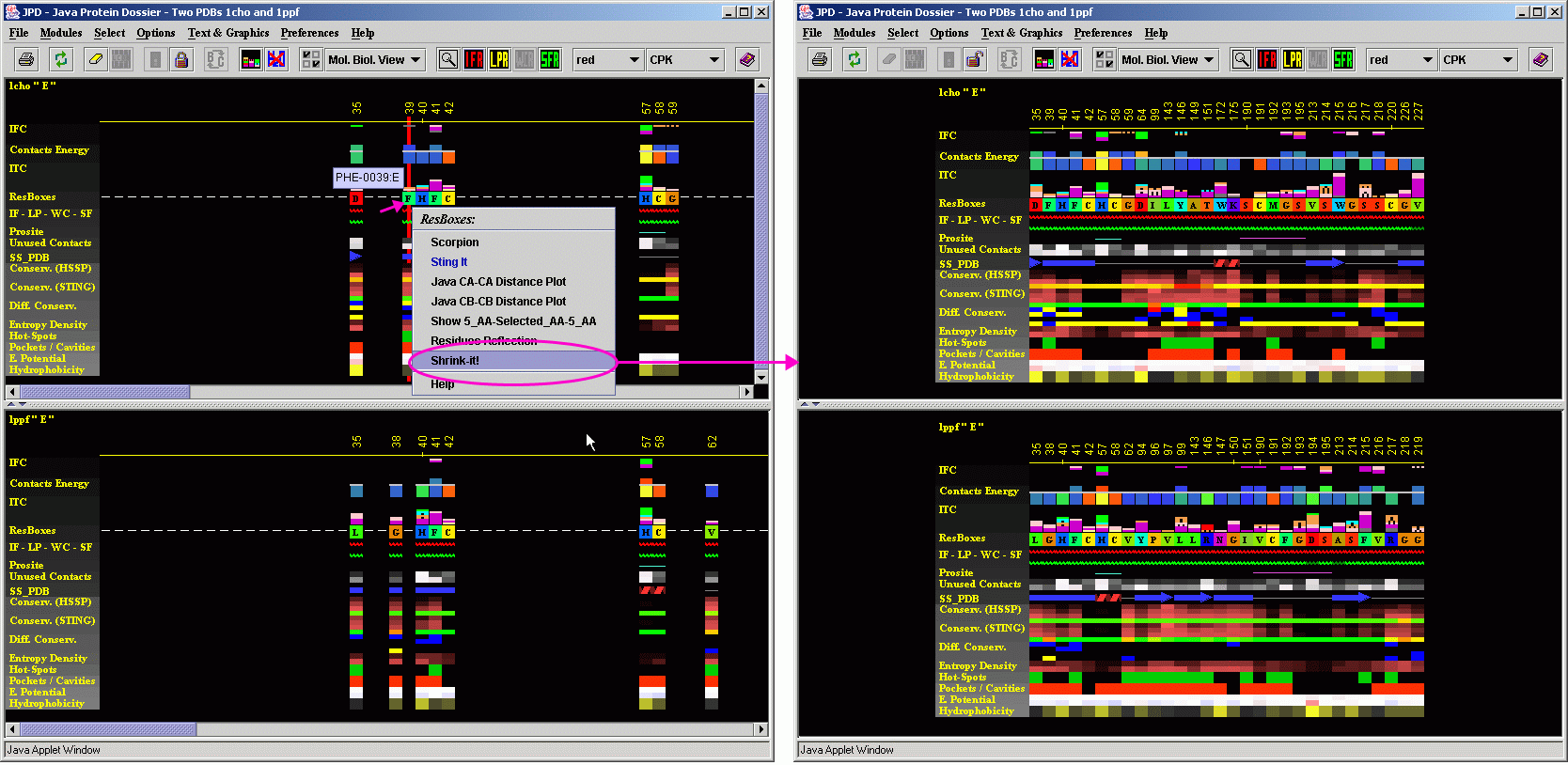
JPD for 2 3D aligned structures
This
is one of the key features that Java Protein Dossier offers to the users
- a possibility to structurally align two proteins and show their sequences
aligned so that they follow structure, rather than sequence alignment.
JPD can handle both two
publicly available (PDB deposited) files or two
local (non public) pdb formatted files.
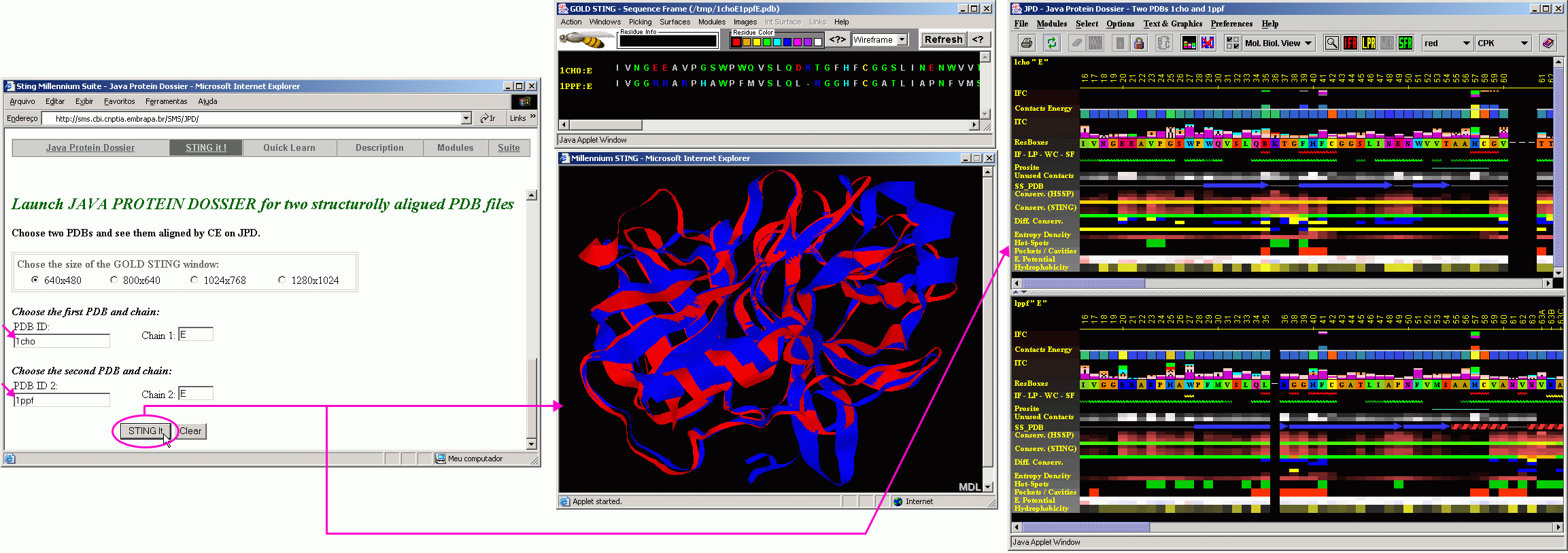
To start JPD in aligned structure mode, a user needs to indicate two PDB files with structures to be aligned. In this "tutorial"case, we used two serino proteases: Chymotrypsine (1cho.pdb) and elastase (1ppf.pdb). As both files contain both enzyme (E) chain and an inhibitor (I) chain, we need to indicate which chains are to be aligned structurally. In this case we asked that the alignment is done on E chains. Alignment is performed by the CE software within BLUE STAR STING environment.
As a result, BLUE STAR STING will show in CHIME window, two aligned structures and in the BLUE STAR STING sequence window, the sequences of two chains, aligned according to the structural alignment. JPD will show parameters of both chains (first chain/structure in the upper and the second chain/structure in the lower JPD window) also following a structural alignment (with gaps introduced to satisfy optimal structural alignment). A user can then access the STING Components of either structure by clicking with the right mouse button at the corresponding parameter line and selecting from resulting menu the component to be initialized. At this moment, the JPD menu items: Modules, would not work properly for aligned structures, so a user is kindly asked to use alternative path to call the STING components as described above).
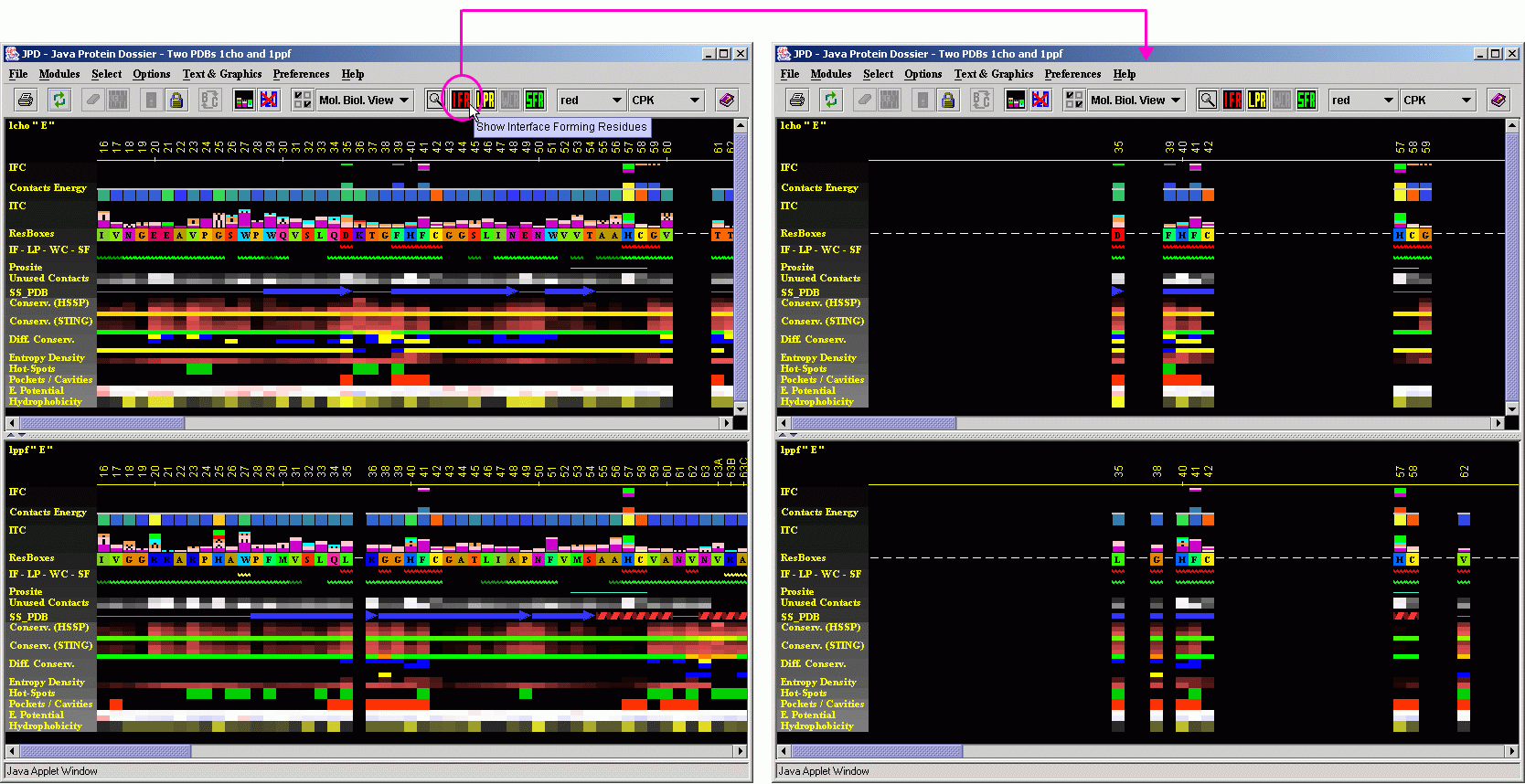
A most useful option that JPD offers in structural alignment
mode is the one which permits the user to see information on INTERFACE
residues. The user should click on the IFR icon and observe JPD showing
only the residues at the interface area of two chains. It is necessary
to say that the IFR residues (underlined by the red wiggly line) for each
of the shown chains, are calculated for the complex of the E chain with
the I chain for respective PDB files. In addition, JPD will preserve alignment
mode, so that a user may do a quick inspection of the spatial position
of those important "interfacing" residues. This clearly offers
a quick guide to the mechanism of the specificity for certain substrates/inhibitors
practiced by proteins under the analysis.
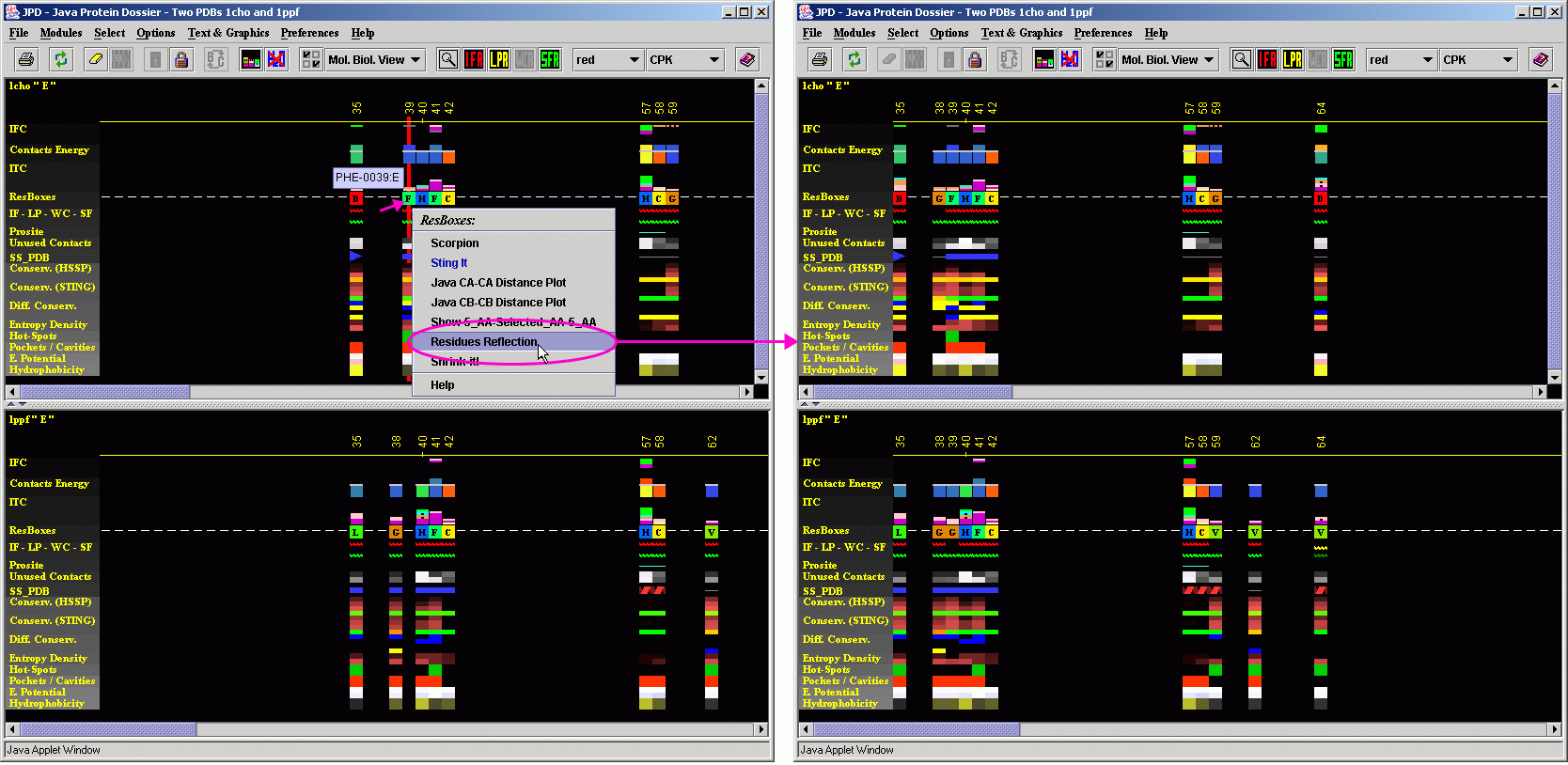
A user may also compare the residues which are possibly
not at the IFR area, but should be inspected anyway because they occupy
structurally identical position with the one in the structurally aligned
protein, where equivalent position is identified as the one at the Interface
area. To do this, a user should use the menu option "Residue Reflection".
Compare obtained image with the one on the figure above. Here, JPD "reflects"
as in mirror image, all residues which are at the interface, making all
of them to have a pair on the aligned structure (except for the gaps,
where such reflection is impossible - see position of Valine_62 in 1PPF).
Finally, for publication purposes, a user may "shrink"
the JPD image and eliminate the gaps and spaces separating IFR residues,
creating effectively an image of the amino acid IFR ensemble.